By Kylie Ryan Kaler
Over the years, the landscape of drug development in the United States has undergone a significant evolution, transitioning from an unregulated consumer market to a heavily regulated industry. This pivotal shift has compelled drug developers to shoulder the responsibility of establishing the safety and efficacy of their drug products. In the United States, the number of companies manufacturing drug products and the total number of products has greatly increased over the years. This increase necessitates the enforcement of safe and reliable practices to protect the consumer. Regulations provide structure that guides the pharmaceutical companies through drug discovery, which results in drug products that are both safe and effective for a given disease. Dan Gordin, Ph.D., led a detailed workshop with Rutgers iJOBS regarding the history and regulation of drug products.
Prior to 1906, before the advent of stringent regulations, consumers bore the burden of accessing the safety and efficacy of a drug, as there were no regulatory frameworks in place for such evaluations. Consequently, drug developers could market products such as “snake oils” to consumers, which at best, provided marginal relief and, at worst, caused harm to the consumers. In 1906, the First Consumer Protection law was enacted, leading to the establishment of the Bureau of Chemistry, now known as the Food and Drug Administration (FDA). This legislation mandated that drugs must carry labels listing their components and served as a crucial step in ensuring patient’s safety.
In 1937, S.E. Massengill Company created a new formulation of the drug sulfanilamide in ethylene glycol (an antifreeze agent) that was widely used to treat streptococcal infections. This formulation resulted in over 100 deaths across the United States. You can learn more about the sulfanilamide disaster in an article published by the FDA. The public outcry following the sulfanilamide tragedy prompted the passing of the Federal Food, Drug, and Cosmetic Act (FD&C Act), shifting the responsibility of demonstrating safety onto drug developers. In the 1950s, yet another issue arose with the prescription of thalidomide to treat pregnancy-related morning sickness in Germany and Australia. This further highlighted the global impact and need of establishing drug safety. Despite attempts to market thalidomide in the United States, rigorous safety evaluations revealed insufficient data and a lack of demonstrated efficacy. This proactive decision to halt thalidomide marketing prevented severe birth defects and other health conditions associated with thalidomide, including neuropathy, deep vein thrombosis, and pulmonary embolism. This prompted the Kefauver-Harris Amendment to the FD&C Act in 1962, which further reinforced the responsibility of proving safety and effectiveness on the producer and initiated the regulation of clinical trials for drug discovery. While the regulatory landscape shifted the burden of drug safety from consumers to manufacturers during the 1900’s, unfortunately, many consumer lives were lost during the absence of robust regulations.
In its current state, the FDA’s mission is to protect public health by regulating the development of food and drugs to ensure both safety and efficacy. The FDA regulates approximately 25% of the United States’ market, which includes various sectors such as food, pharmaceuticals, cosmetics, medical devices, over the counter medicines, veterinary medicines, and tobacco products. With an annual budget of $6.5 billion, the FDA employs approximately 18,000 scientists, reviewers, auditors, and policy makers. To safeguard its mission, the FDA relies on the FD&C Act and an extensive code of federal regulations, which delineate the procedures and requirements for introducing a new drug (using an investigational new drug application) or a medical device (facilitated through either the premarket approval application or demonstrating equivalency to an already existing device). It is important to note that the FDA does not conduct clinical studies; rather, it assumes the role of meticulously reviewing them. The agency looks at studies for data anomalies and/or assesses the weaknesses of the study design or statistical analyses, then decides the level of safety and efficacy of the drug or medical device.
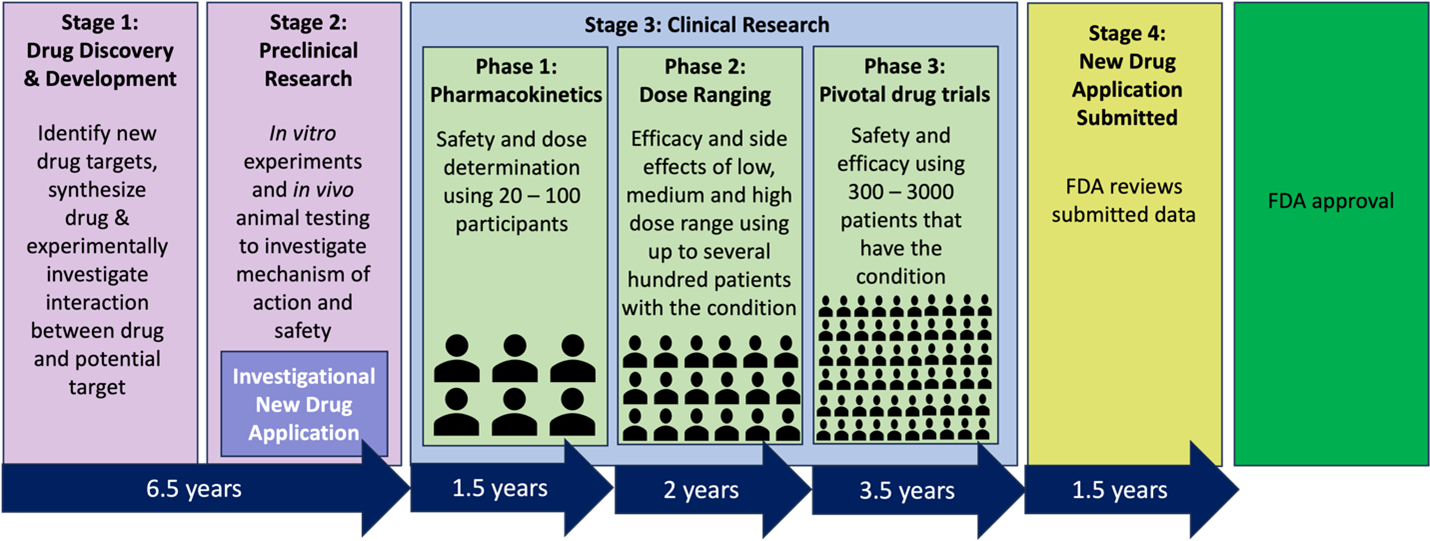
The journey of an experimental drug takes 12-15 years from the lab to patients in the United States. On average, only 5 out of 5000 compounds that undergo preclinical testing progress to human testing, and only 1 out of these 5 is ultimately approved by the FDA. The preclinical or discovery period of research takes approximately 6.5 years and is dedicated to assessing the safety, biological activity, and formulation of the compound (Figure 1: stage 1). This stage consists of identifying potential receptors for a new drug, synthesizing the new drug, and experimentally investigating the interaction between the drug and its anticipated target. Subsequently, the new drug then undergoes animal testing to understand its safety profile and mechanisms of action (Figure 1: stage 2). After extensive experimentation, a promising compound may be submitted to the FDA via an Investigational New Drug (IND) application. An IND application allows for the yet to be FDA-approved drug to be distributed across state lines for clinical investigation. Once approval has been granted, the compound advances to clinical trials. Phase I clinical trials focus on determining the safety and dosage of the drug candidate with a sample size of 20 – 100 healthy human patients and lasts approximately 1.5 years. Phase II clinical trials, a 2-year process, aims to determine the effectiveness and identify potential side effects of the drug candidate using a larger pool of 100 – 500 human patients. Phase III clinical trials further evaluates effectiveness and monitors for adverse reactions during long-term use within a population of 1000 – 5000 patients. Phase III trials must consist of two pivotal studies showing duplicate outcomes and generally takes 3.5 years to complete. The cost of drug development escalates greatly as the drug candidate progresses from Phase I through Phase III. Post clinical trials, the New Drug Application (NDA) is submitted to the FDA for review and an approval process that takes approximately 1.5 years. Once the drug candidate enters the market, both the FDA and the drug producer actively monitor its safety.
Once the NDA is submitted, the FDA undertakes a critical assessment of the benefits and risks associated with the new drug. This evaluation considers the severity of the targeted disease, the comparative advantages of the new drug versus the disease standard-of-care treatment, the risk level of the new drug relative to the disease itself, and the availability of any safer alternatives. For example, the previously mentioned drug thalidomide was not approved for use in pregnancy-related morning sickness in the United States in the 1950’s. It was, however, approved for treating multiple myeloma (a cancer of the blood) in 2006. In this case, the seriousness of multiple myeloma outweighed the risks (fetal toxicity, neuropathy, deep vein thrombosis, pulmonary embolism) of treatment with thalidomide.
What was once an adversarial relationship between drug producers and the now termed FDA has transformed into a partnership that enables the development of safe and effective drugs to enhance human health. This collaboration has played a pivotal role in increasing the average human lifespan and overall quality of life increasing over the decades in the United States.
This article was edited by Senior Editor Sonal Gahlawat and Senior Editor Natalie Losada.